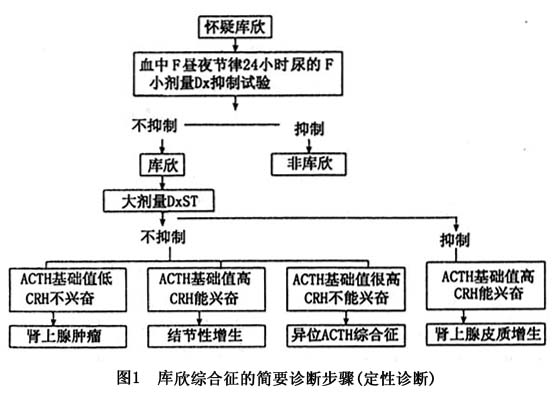
发病机制
发病机制
发病机制:
1.原发性肾上腺皮质病变 原发于肾上腺本身的肿瘤,其中皮质腺瘤约占成人库欣综合征的20%。皮质腺癌约占5%;而在儿童,50%以上的腺瘤是恶性的。肾上腺肿瘤的生长与分泌功能有自主性,不受垂体分泌的ACTH的控制,故称非ACTH依赖型。由于肿瘤分泌过多的皮质醇,反馈抑制了垂体ACTH的释放,病人血中的ACTH很低,以致对侧与同侧瘤外的肾上腺皮质萎缩。腺癌一般较大,生长迅速,除分泌过多的皮质醇外,还分泌其他皮质激素(醛固酮)及肾上腺雄激素;腺癌患者可有11β-羟化酶受累,病人尿中的17-酮类固醇(17-ks)显著增高。肾上腺腺癌的患者都有17号染色体长臂上的p53基因的突变。p53基因是一肿瘤抑制基因,它能起免疫监视作用,使机体及时清除突变的细胞株。而当p53基因突变或缺失,则免疫监视作用就丧失,突变的细胞株就能无限繁殖,产生肾上腺皮质的恶性肿瘤。而在肾上腺良性肿瘤中,未发现p53基因的问题。故良性肿瘤的机制还未明确。
还有专家认为,肿瘤的性质与11号染色体短臂的缺损有关;大多数恶性肿瘤有11号染色体短臂缺损,可导致。IGFⅡ的过度表达,使异型细胞过度生长。
2.垂体瘤或下丘脑-垂体功能紊乱 继发于垂体瘤或下丘脑-垂体功能紊乱的双侧肾上腺皮质增生,称库欣病,约占成人库欣综合征的70%。由于垂体瘤或下丘脑-垂体的功能紊乱,分泌ACTH过多,刺激双侧肾上腺皮质增生,可引起皮质醇过多分泌,故属ACTH依赖型。
不论垂体有无肿瘤,ACTH细胞分泌ACTH均受下丘脑中ACTH释放激素(CRH)的调节;CRH能促进ACTH的合成与释放,而神经递质对ACIH的分泌也有影响,血清素(5-羟色胺)及乙酰胆碱(Ach)可对ACTH的分泌起兴奋作用,而多巴胺(DA)和去甲肾上腺素(NA)则起抑制作用。这些神经递质可能通过兴奋或抑制CRH的分泌来起作用。中枢神经递质的紊乱会导致ACTH分泌过多,从而引起肾上腺皮质增生。下丘脑-垂体的功能紊乱,有时也会累及腺垂体其他激素,如泌乳素(PRL)与生长激素(GH)等。
除了下丘脑-垂体功能紊乱引起的库欣病外,更多的是由于垂体腺瘤,占库欣病患者的80%~96%。此组患者中有腺垂体大腺瘤(直径>10mm)伴蝶鞍扩大者约占10%,近来开展经蝶窦垂体显微手术的越来越多,已证实蝶鞍不扩大者80%以上有垂体微腺瘤(直径<10mm)的存在。这些产生ACIH的腺瘤细胞大多为嗜碱性或嫌色性的,实际上,这些嫌色性细胞具有旺盛的分泌功能,可能是由于分泌颗粒在形成后迅速释放,故表现有嫌色性。
最近的资料显示,大多数垂体微腺瘤是单克隆的,可自主地分泌ACTH,并促进双侧肾上腺增生使血皮质醇增多,而增多的皮质醇又能长时间抑制CRH的释放,使垂体微腺瘤旁正常的ACTH细胞萎缩。库欣病患者于垂体瘤摘除后,可出现暂时性肾上腺皮质功能低下,而且病理上可发现ACTH瘤周围的正常垂体组织仍处于静止状态,这一点与肾上腺腺瘤相似。以后库欣病的症状与生化指标会明显好转,ACTH及皮质醇的昼夜节律能逐渐恢复,并无永久性的下丘脑功能紊乱。
也有人认为:增生型皮质醇增多症中,垂体分泌过多的ACTH并非完全是自主性的(即使有明显垂体肿瘤者也如此),而是受到下丘脑及其他中枢的影响。大剂量(8mg)的地塞米松能抑制ACTH的释放。美替拉酮(肾上腺皮质11-羟化酶阻滞物)可通过减少皮质醇的合成而促进ACTH大量释放,神经垂体的加压素(包括精氨酸加压素与赖氨酸加压素)也具有类似CRH的作用,可促使ACTH释放。神经垂体(神经垂体)素与CRH有协同作用,联合运用能促进ACTH的大量释放。
库欣病患者的双侧肾上腺都是弥漫性增生,病根在垂体或下丘脑。如果对这类患者仅针对肾上腺作双侧肾上腺切除,则原来的垂体微腺瘤缺乏血中皮质醇的负反馈抑制,会逐渐增生,甚至破坏蝶鞍,过度分泌ACTH,血浆中ACTH的水平将会极度增高,造成皮肤色素沉着,称Nelson综合征。过度增大的垂体瘤称Nelson肿瘤。
库欣病患者在大量ACTH的持续兴奋下,可出现双侧肾上腺皮质增生,进一步发展在某些患者中可出现结节,甚至小腺瘤。这种腺瘤往往为多发性的,大小不一。在发展过程中,肾上腺增生性结节的分泌功能可逐步变为自主性的,称结节性增生。
3.异位ACIH综合征及异位CRH综合征 少数病例由于垂体-肾上腺以外的癌肿,产生具有ACTH活性的物质或大分子ACTH(正常ACTH分子质量为4.5 kD,大分子ACTH分子质量为20kD),或具有CRH活性的物质,可刺激垂体及肾上腺分泌过量的皮质醇而发病,属于ACTH依赖型。过去一般认为,异位ACTH综合征时最多见的是肺癌,特别是燕麦细胞癌(约占50%),其次为胸腺癌(约占20%)、胰腺癌(约占15%),其他还有起源于神经嵴组织的肿瘤、甲状腺髓样癌以及消化系统和泌尿系统的癌等。
现在发现,能异位分泌ACTH的肿瘤,有很大一部分是偏良性的肿瘤,如类癌。胸腔的类癌主要是支气管类癌,约占所有异位ACIH综合征的40%,燕麦细胞癌在第2位,占8%~20%,胸腺癌与胰腺癌各约占10%,肝癌、前列腺癌、乳腺癌分占余下的比例。没有中胚层来源的肿瘤如
肉瘤分泌ACTH的报道。
过去之所以不把类癌放在第1位,是因为很多类癌(如支气管类癌)呈“隐性”异位ACTH综合征。所谓“隐性”异位ACTH综合征,Doppman把它定义为非垂体源性的,CRH或ACTH依赖的高皮质醇血症,在4~6个月中没有出现明显肿瘤来源的异位ACTH综合征。“隐性”异位ACTH综合征很容易与库欣病混淆,因为用实验室检查与影像学检查均不能鉴别,易误导,这常导致一些不必要的垂体或肾上腺手术。这就更需要改进鉴别诊断过程,例如测岩下窦静脉血的ACTH与外周血中的ACTH。
必须注意,异位ACTH综合征患者的肿瘤不仅产生ACTH,还分泌其他激素。APUD系统的很多肿瘤能合并异位.ACIH综合征。一些良性肿瘤也能分泌ACIH样物质,如嗜铬细胞瘤能异位分泌ACTH,使血中ACTH的浓度增高,临床表现完全像库欣综合征。特别当嗜铬细胞瘤不在肾上腺时,患者常被作为“垂体性库欣病”而作垂体手术,术后无疗效,只有当检查到24h尿中儿茶酚胺增高,才最后诊断为嗜铬细胞瘤;很容易漏诊。
大多数异位ACTH综合征患者血中的皮质醇不受大剂量地塞米松的抑制,但有30%的隐性异位ACTH综合征患者的高皮质醇能被地塞米松所抑制。41%的胸腺类癌,其隐性异位ACTH综合征能被大剂量的地塞米松抑制。另外也有9%~25%的垂体性库欣综合征患者的皮质醇不能被大剂量地塞米松所抑制,这样就特别容易混淆。
异位ACTH综合征有一些肿瘤标志物:
(1)大分子质量的ACTH:测定ACTH前体物质厂ACTH的比例,在异位ACTH中为58∶1,而在库欣病中为5∶1,这对异位ACTH综合征的诊断很有帮助。
(2)降钙素。
(3)肠道激素(如胃泌素、胃泌素释放肽)。
(4)癌胚标志物(如CEA,AFP)。
(5)胎盘标志物(hCG,β-HCG)。
(6)5-羟吲哚乙酸(5-HIAA)。
(7)APUD标志物(α-烯醇化酶、嗜铬粒蛋白)。
90%以上的隐性异位ACTH综合征包含
神经内分泌肿瘤,绝大多数病例都能测到α-烯醇化酶和(或)嗜铬粒蛋白。
异位CRH综合征极少见,常伴随着异位ACTH综合征。该类患者常常不被CRH兴奋,不受大小剂量的地塞米松抑制,而且肿瘤(恶性)发展快,原发癌肿的症状很明显。
曾有人报道一种胸部肿瘤,不分泌ACTH或CRH样物质,而分泌铃蟾肽(bombesin)样肽。这类物质能提高CRH的生物活性,在垂体水平上致ACTH高分泌。
4.不依赖ACTH的肾上腺结节性增生(或称结节性发育异常) 近年报道少数患者呈现双侧性肾上腺结节性增生,但并非由于ACTH过多所致。其中又可分为两型:一型见于中年人,肾上
腺病变呈大结节性;另一型见于年轻者,病变呈深色小结节性,肾上腺有色素沉着,后者常为家族性的。该类患者的病因不详,为ACTH非依赖型,有人称为“原发性增生”,可能有某种ACTH以外的物质刺激肾上腺而引起增生。有人认为是由于产生了兴奋性免疫球蛋白引起的,就像Graves病
甲亢有甲状腺兴奋性抗体那样。
5.医源性类库欣综合征 由于长期应用大剂量的糖皮质激素治疗某些疾病,引起医源性的血中皮质醇增高,患者的临床症状类似库欣综合征,但其本身的垂体-肾上腺皮质受到抑制,功能减退;一旦突然停药,或在应激情况下,可引起急性肾上腺皮质功能衰竭。
临床表现
临床表现:常见的临床表现有:
1.特殊体征 患者呈向心性
肥胖、满月脸、水牛背,皮肤菲薄多血质,体表毳毛增多,头发多油脂,面部、前胸后背部痤疮,在大腿外侧、臀部及腹壁两侧可见宽大的紫纹。
2.高血压、低血钾 皮质激素可使钠回吸收增加,总体钠含量升高,血容量扩大,血压升高伴轻度水肿,尿钾排出增多而引起低血钾、轻度碱中毒。
3.
糖尿病或糖耐量异常 由于皮质激素促进糖原异生,拮抗胰岛素的外周效应,多数皮质醇增多症者可有
糖尿病或糖耐量低减。
4.骨骼、肌肉代谢异常 由于皮质醇促进蛋白质分解,同时减少其合成,本病患者可长期处于负氮平衡,而表现为肌无力,肌萎缩,皮肤变薄,毛细血管脆性增加而易发生瘀斑,骨质疏松导致腰背疼,并可发生病理性骨折。儿童可出现生长发育停滞,青春期延迟等。
5.性功能紊乱 女性可出现月经紊乱或
闭经,男性有
阳痿。
6.易感染 皮质醇增多症患者抵抗力下降,易患各种感染如细菌、真菌感染,伤口不易愈合。
7.精神症状 患者有
失眠,情绪不稳,注意力不能集中。少数患者还可有躁狂、抑郁或
精神分裂症。
8.高尿钙与肾结石 皮质醇可促进骨钙释出以及抑制肠道钙吸收,促进尿钙排量增加,故易发生泌尿系结石。
其他辅助检查
其他辅助检查:
1.X线检查
(1)蝶鞍平片法或分层摄片法:由于库欣病患者的垂体肿瘤较小,平片法结果大多阴性,用蝶鞍分层片法部分病人仅有轻度的异常改变,且敏感度差,准确性不大。但如发现蝶鞍增大,有助于垂体瘤的诊断。
(2)肾上腺X线法:对肾上腺占位性病变的定位有帮助,但不能鉴别结节性增生与腺瘤。
2.CT检查 由于CT扫描的每1层约10mm,对于直径>10mm的垂体腺瘤,CT的分辨率良好,但对直径<10mm的垂体微腺瘤,CT有可能遗漏,阳性率可达60%。所以CT。未发现垂体瘤者,不能排除微腺瘤的可能。
对肾上腺增生与腺瘤的检查,CT的作用大,分辨率好,因为肾上腺腺瘤的直径往往>2cm。
注意:CT检查,要注射造影剂,为了防止变态反应,一般都给予10mg地塞米松;CT检查要安排在大剂量的地塞米松抑制试验以后,否则要间隔7天以上再做大剂量的地塞米松抑制试验。
3.磁共振(MRI)检查 对库欣病,MRI是首选方法,与CT相比可较好地分辨下丘脑垂体及鞍旁结构(海绵窦、垂体柄和视交叉),但对直径<5mm的肿瘤,分辨率仍仅为50%。
4.B超 对肾上腺增生与腺瘤好,属无创伤检查,方便、价廉、较准确。常用来与MRI,CT一起作库欣综合征的定位诊断。
5.其他
(1)131Ⅰ-α-碘化胆固醇肾上腺扫描:能显示肾上腺腺瘤的部位和功能;腺瘤侧浓集,对侧往往不显影,图像不如CT清晰。
(2)岩下窦ACTH测定(IPSS):做选择性静脉取血,测ACTH。若病人经生化检查为库欣病,而CT等扫描为阴性,可做此检查。
从岩下窦(垂体的主要静脉流出通道)、颈静脉球及其他部位取血测ACTH,与末梢血中的ACTH比较:库欣病患者患侧岩下窦的血中ACTH与末梢血中ACTH的比值多≥2∶1;异位ACTH综合征测岩下窦的血与末梢血中的ACTH不会有梯度改变(一般≤1.5∶1);若一侧岩下窦血中ACTH的水平与对侧相比≥1.4,说明垂体腺瘤局限于这一侧。另外选择性静脉取血查ACTH,还可判定可疑肿瘤部位是否有异位的ACTH分泌。
双侧岩下窦取血(IPSS)如结合CRH试验,可使诊断的精确性达到100%。Finding等认为,垂体性库欣病患者,其岩下窦与外周血中基础ACTH之比为11.7±4.4,在应用CRH后可增至50.8±18.3,而在异位ACTH综合征病人中,IPSS与外周血中基础ACTH之比为1.2±0.1,应用CRH后无变化。这在区分隐性ACIH综合征患者时特别需要。
治疗
治疗:
1.手术 肾上腺肿瘤可手术切除,但对垂体ACTH分泌肿瘤引起双侧肾上腺增生症,过去多采用一侧全切,另一侧次全切除法,然后对垂体行放疗。现主张经鼻垂体瘤切除。
2.垂体放疗 可与手术治疗联合应用。
3.药物 主要作用于肾上腺抑制皮质醇合成,密妥坦6~10g/d,分次口服;氨鲁米特(氨基导眠能)0.75~1.0g/d,分次口服;甲吡明1.0g/d,分次口服;或酮康唑0.2~1.0g/d,分次口服。
4.血清素拮抗剂 作用于下丘脑垂体,抑制皮质醇合成,如赛庚啶24mg/d,分次口服。
5.对症治疗 降血压,降血糖,补钙等,非甾体抗炎药可缓解骨疼。